eDNA and other monitoring methods based on molecular biology
With the help of molecular biology, we can improve the coverage, accuracy, and cost-effectiveness of environmental monitoring, enabling a much more complete picture of biodiversity and the state of the environment and their trends.

On this page:
- What are molecular monitoring methods?
- Most commonly used molecular methods
- Possibilities and challenges of the methods
- Standardization and quality control
- Implementation in Finland
- How to implement molecular methods?
- Welcome to the national eDNA idea group
- Resources
What are molecular monitoring methods?
Molecular methods refer to techniques based on DNA or RNA analysis, which can be used in nature monitoring. Based on DNA, a single target species can be detected or even hundreds of species can be identified from individual samples. Population structure and other intra-species variation can also be studied using DNA methods. RNA-based methods produce information about the genes expressed in the studied environment and thus about the biological processes going on in the ecosystem.
Many types of samples can serve as source material for molecular analyses. To verify species identification and to study genetic variation within the species, tissue samples taken directly from the organism, or for example, excrement samples are most often used.
Environmental DNA or eDNA
Environmental DNA or eDNA refers to the DNA contained in samples collected from the environment, for example water, soil, sediment, snow, or air.
Environmental DNA samples are usually used to either identify a specific target species or to find out the species of a certain species group or groups as comprehensively as possible. Also for samples collected with traditional sampling methods, such as fishing, the species can be identified with eDNA methods instead of morphological identification. This is done from a DNA homogenized compilation or a bulk sample or for example, from the liquid used to preserve the sample.
The costs of massive parallel sequencing and other methods, which enable the simultaneous detection of even hundreds of species, have significantly decreased in recent years. Chains of methods, from sampling to interpretation of results, have developed to a degree that enables their large-scale, routine use in environmental monitoring. With the help of new methods, the coverage, accuracy, and cost-effectiveness of monitoring can be improved enabling a much more complete picture of biodiversity and the state of the environment and their trends.
Most commonly used molecular methods
Ensuring species identification of an organism: DNA barcoding
In DNA barcoding, DNA is isolated from a sample taken from an organism and a certain short region suitable for species identification (“barcode”) is sequenced, whose base sequence is compared to a database of confirmed species-specific barcode sequences. In addition to traditional Sanger sequencing, massive parallel sequencing (so-called second and third generation sequencing) is also used in DNA barcoding today.
Identification and abundance determination of individual target species: PCR tests, qPCR, dPCR, ddPCR
In a PCR test only the DNA of the target species is duplicated from the DNA isolated from the sample using a species-specific primer. With quantitative PCR methods (qPCR, dPCR, ddPCR) it is also possible to determine the abundance of DNA of the target species in the sample.
Species determination: metabarcoding (2nd and 3rd generation sequencing)
In metabarcoding, up to hundreds of thousands of sequences of a certain barcode area are produced from one environmental sample by massive parallel sequencing. The sequences can be identified by comparing the DNA base order with the sequence database. The target group of organisms is limited by the barcode area and the PCR primers used in DNA duplication. With metabarcoding, it is possible to find out a large part of the species in that species group in the sample. In addition, the method provides indications of species abundance ratios. The proportion of identified species and the accuracy and reliability of the identifications primarily depends on the quality and comprehensiveness of the reference database used.
In metabarcoding, the so-called second-generation sequencing technology (e.g. Illumina NovaSeq, MiSeq, NextSeq), which requires DNA duplication and sequencing to be limited to a specific gene region of a maximum length of approx. 300 base pairs. The newer so-called third-generation sequencing technology (e.g. Oxford Nanopore, PacBio) can produce much longer sequences (tens or hundreds of thousands of base pairs) from the entire genome area, and DNA duplication is not necessary.
In principle, the new technology makes it possible to reach all groups of organisms with the same analysis and also to examine intra-species variation. In practice, however, the utilization of the method is still limited by the coverage of whole genomes and genome libraries, the fragmentation of DNA obtained from environmental samples into smaller pieces, and to some extent also the insufficient quality of long sequences.
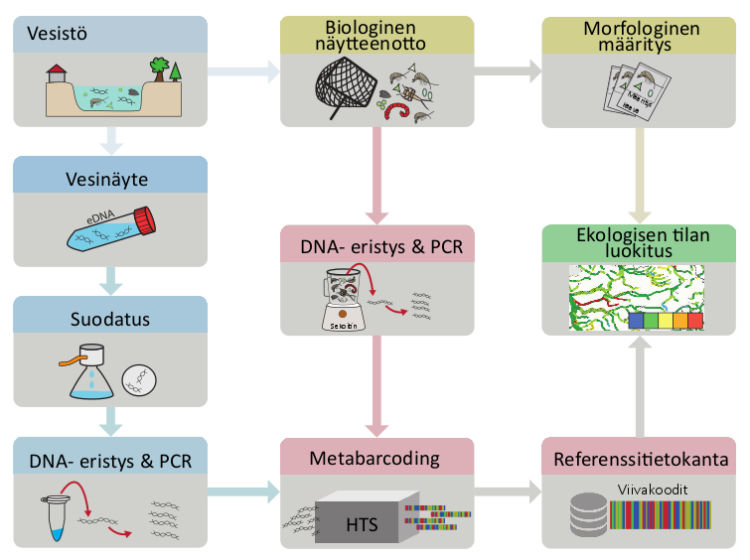
Exploring intra-species variation: sequencing of single genes, genomics, SNP and microsatellite marker genes
With current methods, the investigation of intra-species variation usually requires samples that can be linked either to a single individual or to a specific population. Determining intra-species variation from eDNA samples collected from the environment has been developed in a few research projects, but suitable methods are still unattainable for most species for the time being.
Genetic diversity within a species can be assessed with many different methods, which aim to answer slightly different questions. They also differ in how much background information about the genetic structure of an organism must be available in order for the method to be applied. However, with the decrease in sequencing costs and the development of methods, the collection of genetic background information is no longer a major bottleneck for examining the intraspecific genetic diversity of natural populations.
Sequencing single genes
At its simplest, genetic diversity can be viewed in terms of a single gene. This can be meaningful, for example, in situations where a particular gene is of particular interest, for example because it is known to significantly affect some characteristic of an organism.
The intra-species variation in a single gene region can be assessed either directly by sequencing the gene regions in question from several individuals (in the simplest way using the Sanger method) and by examining the variation in base pair order or structural features of the gene (e.g. insertions, deletions or inversions). By looking at the number of genetic forms determined in this way, various key figures for the diversity of the genetic area can be calculated (e.g. observed number of genetic forms, relative proportions of genetic forms and average heterozygosity of the gene locus).
Genomics
Determining the variation in a single gene does not reliably indicate the amount of variation in other genes or the whole genome. When looking at natural diversity, variation at the level of the entire genome is often more interesting than individual genes, because it is closely linked to the effective population size and thus to the long-term probability of survival.
To determine the variation at the genome level, one must aim to sequence either entire genomes or a sufficient sample of different gene regions of the genome from several individuals and/or populations. Whole genome sequencing is most often performed by sequencing short strands of DNA isolated, digested, and amplified from a sample individual. These short sequences are then assembled into a complete genome using computational tools and already assembled control genomes. Some new technologies also allow longer DNA strands to be sequenced without splitting and duplication steps. Such technologies will possibly become more common in the future, as they rely less on computational solutions and are not as sensitive to errors in the duplication phase. Whatever the method, comparing assembled genomes identifies regions that vary between individuals and thus contribute to the genetic diversity of a species or population.
Genome sampling can also be done in many different ways. In the Restriction site Associated DNA Sequencing (RAD-seq) method, DNA isolated from a sample is enzymatically splitted, which produces DNA strands that represent the genome evenly, but reduces the amount of DNA to be sequenced. RAD-seq enables fast and cost-effective analysis of large populations and many samples, which is often desirable for the assessment of intra-specific genetic diversity.
SNP and microsatellite marker genes
Before the popularization of RAD-seq and whole-genome sequencing-focused methods, it was customary to estimate genome-wide variation using some kind of marker gene set. Typical marker gene sets are the different gene forms observed in individual base pairs produced by point mutations (single nucleotide polymorphism, SNP) or repeat sequences of base pairs (so-called microsatellites). In order for these methods to form a reliable picture of genetic variation, they should be fairly evenly distributed over the entire genome.
Possibilities and challenges of the methods
eDNA and other molecular methods enable species identification that is easily comparable and reproducible internationally. With their help, it is also possible to reach many difficult-to-detect and poorly known groups of organisms that are outside the scope of monitoring based on traditional methods. For example, local water and air samples can be used to reach either individual species or the entire species in a large area, which would not be possible to map with traditional methods due to costs.

Although molecular methods can significantly supplement the overall picture of biodiversity, they do not produce information comparable to methods based on traditional species identification. For example, meta-barcoding does not provide information on the number of individuals of a species or the characteristics of individuals such as age, size or sex. In addition, the information about the relative abundance ratios of species obtained by meta-barcoding is still generally only indicative at the moment. However, methods are constantly being developed in a more quantitative direction.
Another significant challenge is related to the interpretation of environmental DNA data. Since environmental DNA detects DNA present in for example water and not directly living individuals, it is necessary to evaluate on a case-by-case basis what the observations tell about the presence of individuals. The transport of DNA and the rate of degradation in the environment are affected by numerous factors, such as flow conditions, temperature, and sunlight. The degradation and migration of DNA in different environments has been investigated in many scientific studies, the results of which should be taken into account when planning molecular biological monitoring. Various models describing the migration of DNA are also useful in evaluating possible source areas, but the predictions of the models are inevitably always accompanied by uncertainty.
Molecular monitoring methods are currently being actively developed around the world for different groups of organisms and ecosystems, and individual methods have also been put into routine use in several countries. However, the use of molecular methods in monitoring on a large scale is still at an experimental stage, and the field of development projects and expertise is fragmented.
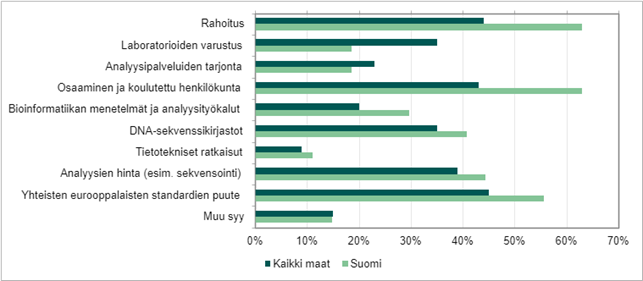
Insufficient funding, lack of skilled workers, gaps in reference libraries especially for certain northern species groups, and the lack of international standards for methods are considered by experts in the field to be the most important factors limiting the introduction of methods. There are national and international methodological guidelines, but they are scattered, and the minimum requirements have not been generally accepted or agreed upon, i.e. there are hardly any real, commonly accepted standards yet. Decentralized funding easily causes guidelines to be developed at the national or organizational level, but there is not enough exchange of information or sharing of lessons learned between projects and different countries. This causes a great risk that the molecular biological data are not comparable at the international level.
Standardization and quality control
International standardization of methods is very important to ensure the quality and comparability of molecular biological data. Standardization is currently still in its early stages, but it is being actively promoted by many different measures in Finland and internationally.
The CEN standard EN 17805:2023 Water quality – Sampling, Capture, and preservation of environmental DNA from water has been published in early 2023. An eDNA-themed working group is being established under ISO/TC 147/SC 5/WG13 “Environmental DNA and RNA methods”. Development manager Kristian Meissner from the Finnish Environmental Insitute oversees the working groups for the already published CEN standard and the ISO standard that is currently under preparation.
The International eDNA Standardization Task Force iESTF (https://iestf.global) was established in connection with the international GEO BON conference in Montreal in October 2023. iESTF cooperates closely with the international research community and various stakeholders.
Recommendations for quality control
We recommend to use existing international standards in all stages of using molecular monitoring methods, from sampling and laboratory analyzes to the analysis of the finished data. When using a commercial service provider, it is a good idea to ensure in advance that the operator commits to high-quality and traceable work in all phases in compliance with existing standards and good laboratory practices. Accurate documentation of the method used is of paramount importance for evaluating the quality and comparability of the data.
Quality control must be considered in all stages of the collection and analysis of molecular biological data, from sampling to analyzes of the finished data using bioinformatics tools. The importance of quality control will increase as molecular environmental monitoring methods are increasingly adopted. Training field samplers, engaging a commercial or research laboratory in quality control that covers all laboratory steps, investing in documentation and traceability of data analysis and storage and training end users in assessing the quality of methods are all very important steps to obtain high-quality molecular biological data.
Implementation in Finland
Molecular monitoring methods are still mostly in the experimental stage in Finland, but the development is fast. In addition to universities, method development and piloting are carried out in all key institutions that coordinate environmental monitoring (Syke, Luke, Central Museum of Natural Sciences, Forestry Agency, Meteorological Institute, Health and Welfare Institute, Food Agency).
The methods are in routine use only for individual game and fish species. The monitoring of entire communities of species using metabarcoding methods has not yet been put into routine use in Finland, but large-scale piloting has been done or is underway, especially with aquatic organisms such as plankton and epiphytic algae, benthic animals, and fish. In the terrestrial environment, large-scale monitoring based on metabarcoding is being piloted, especially with arthropods and fungi.
In Finland, efforts are being made to promote the introduction of molecular monitoring methods, e.g. with research funding and coordination and cooperation between different actors. Molecular methods are included in the Environmental State Monitoring Strategy 2030 published by the Ministry of the Environment, and Syke and Luke have drawn up a road map for the introduction of molecular monitoring methods.

Table: Molecular monitoring schemes in Finland.
| Lajiryhmä | Species/group | System | Methods | Stage | Conducted by | Projects |
|---|---|---|---|---|---|---|
| Yleinen koordinaatio ja datanhallinta | General coordination / data management | Syke, Luke, Luomus, Univ. Oulu, Univ. Jyväskylä, Kuopio | eDNA roadmap, FEO, FinBIF-FIRI2021 | |||
| Virukset | Viruses | terrestrial, freshwater, marine | eDNA metabarcoding, qPCR, eRNA (water, air, wastewater, ticks, fungi, plants, insects) | Pilot | THL, SYKE, FMI, Luke, universities | Wastpan, Finnish Tick Project |
| Bakteerit ja arkit | Bacteria and Archaea | terrestrial, freshwater, marine | eDNA metabarcoding, qPCR (soil, water, air, wastewater, ticks) | Pilot | Luke, SYKE, FMI, THL, RV, universities | MiDAS, Wastpan, Valse V, LUCAS, RECIPE, Finnish Tick Project |
| Kasvien sisällä kasvavat mikrobit | Endophytic microbes | terrestrial | eDNA metabarcoding of bacteria and fungi within plant tissues | Pilot | Luke | |
| Pohjan piilevät | Benthic diatoms | freshwater | eDNA metabarcoding (biofilm, sediment, water) | Pilot | SYKE / Swedish Univ. of Agricultural Sciences | MaaMet, eDNA-monitor |
| Kasviplankton | Phytoplankton | freshwater, marine | eDNA metabarcoding | Pilot | SYKE | GeMeKa, MiDAS, eDNA-monitor |
| Maksasammalet | Liverworts | terrestrial | Bulk DNA metabarcoding | Pilot | Univ. Turku, MH, SYKE | |
| Putkilokasvit | Vascular plants | terrestrial | eDNA metabarcoding/metagenomics (airborne pollen) | Pilot | FMI | |
| Sienet | Fungi | terrestrial, freshwater | eDNA metabarcoding/metagenomics (soil, water, air, litter) | Pilot | Luke, SYKE, FMI, RV, universities | LIFEPLAN, Vesihomehanke, Valse V, RECIPE |
| Jokihelmisimpukka | Freshwater pearl mussel (EN) | freshwater | eDNA + specific PCR | Pilot | Univ. Jyväskylä, MH | SALMUS |
| Pohjaeläimet | Benthic macroinvertebrates | freshwater, marine | eDNA / bulk DNA metabarcoding | Pilot | SYKE | SCANDNAnet, TIMED, MaaMet, eDNA-monitor |
| Maaperän selkärangattomat | Soil invertebrates | terrestrial | eDNA metabarcoding | Pilot | Luke | Valse V, LUCAS |
| Eläinplankton | Zooplankton | freshwater, marine | eDNA, DNA metabarcoding | Pilot | Syke | eDNA-monitor |
| Niveljalkaiset | Arthropods | terrestrial | Bulk DNA metabarcoding | Pilot | Universities | LIFEPLAN, Finnish Tick Project |
| Täpläverkkoperhonen | Glanville fritillary butterfly (EN) | terrestrial | 240 SNP panel, whole genome re-sequencing | Pilot (long-term research) | Univ. Helsinki | |
| Jokirapu, täplärapu | Noble crayfish (EN), signal crayfish (IAS) | freshwater | eDNA + dPCR | Pilot | Luke, Univ. Eastern Finland | |
| Itämeren lohi | Atlantic salmon (VU), Baltic salmon (VU) | freshwater, marine | An array of 220k SNPs | Routine | Univ. Helsinki, Luke | |
| Kalat | Fish | freshwater, marine (coastal) | eDNA + qPCR, eDNA metabarcoding | Pilot | Luke, MMM | SOTKA |
| Sammakko, viitasammakko | Common frog, moor frog | freshwater | eDNA + qPCR | Pilot | Luke, Luomus, MMM | SOTKA |
| Kiljuhanhi | Lesser white fronted goose (CR) | freshwater | eDNA + Sanger sequencing | Pilot | Kiljuhanhi LIFE, MH, Univ. Oulu | |
| Lepakot | Bats | terrestrial | DNA from feces + metabarcoding | Pilot | Luomus | Papanapankki |
| Karhu | Brown bear (NT) | terrestrial | DNA from feces + 96 Single Nucleotide Polymorphism (SNP) panel | Pilot | Luke | |
| Euroopanmajava, kanadanmajava | European beaver (NT), Canadian beaver (IAS) | terrestrial | eDNA (wood chips) + PCR assays | Routine | Luke | |
| Ilves | European lynx | terrestrial | DNA from feces + 96 SNP panel | Pilot | Luke | |
| Valkohäntäkauris | White-tailed deer (IAS) | terrestrial | DNA from feces + microsatellites | Pilot | Luke | |
| Susi, koirasusi | Wolf (EN), wolf-dog hybrids | terrestrial | DNA from feces/urine + 96 SNP panel | Routine | Luke | Susiseuranta |
| Ahma | Wolverine (EN) | terrestrial | 14 microsatellites and mtDNA control region (579 bp) | Pilot | Univ. Oulu, Luke |
How to implement molecular methods?
We have compiled answers to concrete questions that arise for those interested in the use of molecular monitoring methods. If you can’t find an answer to the question that is bothering you, please contact us and we will try to help.
Have molecular monitoring methods been developed for the group of organisms I am studying?
At the international level, the fastest way to find an up-to-date answer is to search for scientific articles. Organism groups whose molecular monitoring is being piloted in Finland are summarized in the table above.
Which method is suitable for my needs?
The chosen method always depends on the research question. For example, the qPCR method is best suited for detecting a single species, while the benthic community of a certain rapid site should be mapped using the benthic metabarcode coding method.
Good guides to support planning are available (see e.g. Bruce et al. 2021 and Pawlowski et al. 2020). The latest information on the technical details of the methods (e.g. the best primers for amplifying the DNA of a certain group of organisms) can be found in scientific articles.
How reliable is the information produced by molecular methods?
The reliability of the results depends on a lot of factors, starting from high-quality sampling to DNA isolation, amplification, sequencing, and analysis of the sequence data with a bioinformatics tool. The methods are developing, and among other things, the species libraries are being supplemented all the time. Please make sure that the sampling has been carefully planned and carried out, that enough samples have been taken in relation to the environment under study (see e.g. Pawloski et al. 2020) and that factors affecting the results such as DNA degradation and migration have been taken into account and the risk of incorrect results caused by them has already been minimized when planning sampling. Also, laboratory quality control in all stages of DNA sample processing and checking the quality of the finished sequence material is of paramount importance to obtain reliable results.
What does the use of the methods require (infrastructure, know-how)?
Nowadays, ready-to-use services are already available, whereby all steps from sampling to analyzing the finished sequence data with bioinformatics tools can be purchased from a commercial operator. Service providers are listed below. A commonly used option is also to do the sampling, DNA isolation and analysis of the finished sequence data yourself and to buy the sequencing of the samples from a commercial service provider.
The introduction of the Oxford Nanopore MINion sequencer to the market has recently revolutionized the field, because due to its affordable purchase price, more and more research groups have had the opportunity to acquire the MINion for their use. However, bioinformatics analysis of sequence data requires special expertise, which has been identified as one of the biggest bottlenecks in several recent surveys.
How can I publish my DNA data?
The Finnish Environment Institute encourages making data openly available as widely as possible. Many international sequence databases and, for example, the international species database GBIF offer a simple way to publish data. Databases are listed under.
In Finland, national solutions for managing DNA-based nature information are currently being developed. When publishing data, the aspects and limitations related to sensitivity must be taken into account (especially human DNA and observations of sensitive species). The international Nagoya protocol(you are switching to another service) regulates the publication of DNA data collected in other countries to ensure international justice in the utilization of genetic resources.
Do DNA methods make traditional taxonomic work unnecessary?
In general, the current view of the scientific community is that DNA methods complement and considerably expand the biodiversity data produced by traditional monitoring methods, but do not replace them. Along with the introduction of molecular methods, traditional methods should also be maintained in the coming years to accumulate reference data and to ensure the continuity of long timeseries. Taxonomic groundwork is the prerequisite for molecular monitoring methods also in the future. The pressure caused by the development of DNA methods should preferably speed up rather than slow down this groundwork.
Is citizen science utilized in eDNA projects? Can I participate?
The BatLab Finland research group of the Central Museum of Natural Sciences (LUOMUS) maps the diets of Finnish bats by inviting citizens to deposit excrement in the excrement bank(you are switching to another service).
Natural Resources Institute Finland (Luke) performs species identification of beavers using beaver food chips. Chips are especially wanted from Pirkanmaa and Western Lapland. More information on Luke website.(you are switching to another service)
Welcome to the national eDNA idea group
The national eDNA idea group was founded in the beginning of 2020. The group aims to provide information on molecular monitoring methods and new projects, upcoming events and other current topics. The group mailing list consists of over 110 people interested on the topic from national research institutes, universities and companies. The group gets together 2-4 times a year by Teams. To join the mailing list, please contact the group leader, researcher Tiina Laamanen (firstname.lastname@syke.fi).
Resources
Guides, methodology instructions and standards
DNA-based species observations
Projects and networks
Hankkeita on listattu eliöryhmäkohtaisesti myös yllä olevassa taulukossa [Käyttöönotto Suomessa]
Service providers
We compile and maintain a list of Finnish companies and other organizations that offer services related to the use of molecular biological monitoring methods. The list is constantly updated – contact us if you would like the service provider you represent to be on the list! The service providers have been verified and have given their consent to the listing, but the quality of the services has not been evaluated for this listing. The listing therefore does not mean a special recommendation by the Finnish Environment Institute. The listing is made in alphabetical order.
More information
Veera Norros
Tiina Laamanen
firstname.lastname@syke.fi